Conformité réglementaire pour le Canada
Conformité réglementaire pour Taïwan
Le ministère taïwanais de la santé et du bien-être (MOHW) est le ministère gouvernemental responsable de la réglementation des soins de santé dans le pays. Il a pour mission d'améliorer la qualité des soins de santé, de prévenir et de contrôler les maladies infectieuses, de garantir la sécurité des aliments et des médicaments et de faciliter le développement technologique.
Au sein du ministère de la santé et des affaires sociales, la Taiwan Food and Drug Administration (TFDA) gère le système réglementaire relatif à la sécurité et à la qualité des aliments, des médicaments, des dispositifs médicaux et des cosmétiques. La TFDA rédige et met en œuvre les réglementations, accorde l'enregistrement des produits et l'approbation des essais cliniques, contrôle la fabrication et l'importation et mène des activités de surveillance de la sécurité des produits de santé.
En mai 2021, Taïwan a annoncé une nouvelleloi sur les dispositifs médicaux , qui modifie la classification des dispositifs médicaux, regroupe les produits par risque et par catégorie, et ajuste les frais d'enregistrement. Cette loi renforce également la surveillance post-commercialisation de la sécurité et de la qualité des dispositifs en imposant des règles pour les rappels de produits.

Le rôle d'un agent taïwanais dans l'enregistrement des dispositifs médicaux
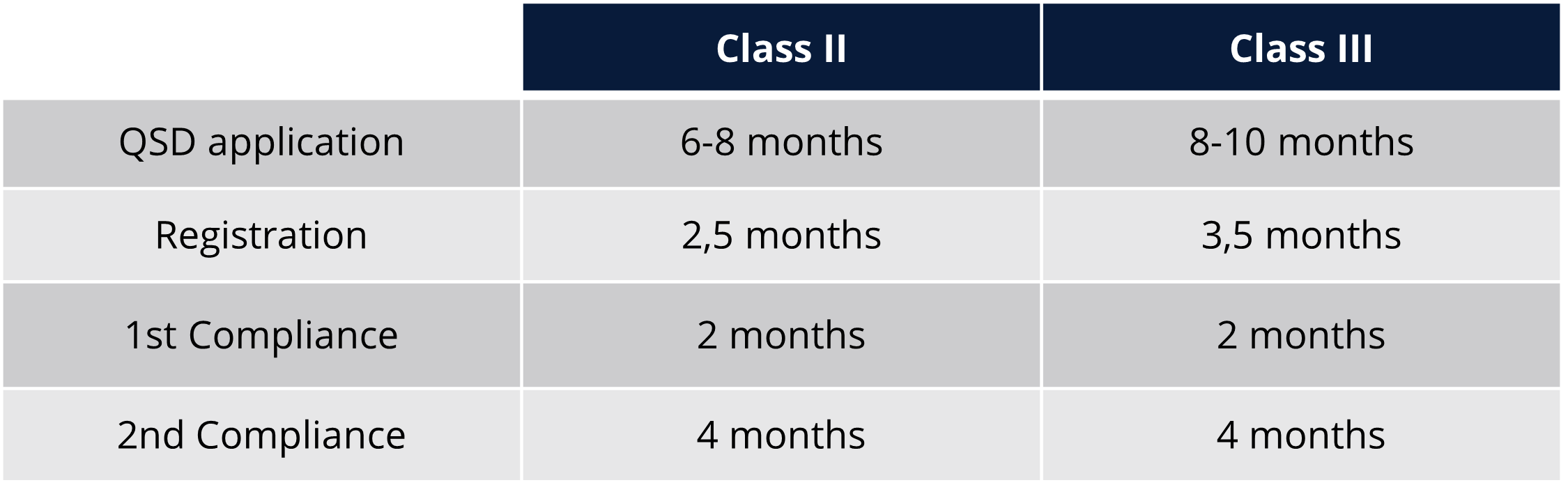
Délais et frais réglementaires pour Taïwan
Classe I (non stérile)
- Les dispositifs médicaux de classe I sans nom de marque (par exemple, les instruments chirurgicaux ou les accessoires électriques) suivent généralement un processus d'autodéclaration simple à Taïwan.
- Délai : Le processus d'approbation de l'enregistrement de la classe I prend environ 3 mois.
- Uneliste de dispositifs declasse I peut être enregistrée par le biais de la procédure d'inscription décrite à l'article 25 de la loi sur les dispositifs médicaux (Medical Devices Act), à compter du 1er octobre 2021.La liste contient 68 codes de dispositifs.
Liens importants et documents d'orientation