
Nous acceptons souvent les effets secondaires des médicaments, mais comment sont-ils gérés pour les dispositifs médicaux selon le règlement européen (MDR), et qu’est-ce que cela implique pour les fabricants?

Blog
Que sont les effets secondaires et quelle est leur relation avec le risque résiduel et les événements indésirables?
Nous connaissons tous les listes d'effets secondaires figurant sur les notices des médicaments et nous les prenons souvent pour acquis. Un effet secondaire est un résultat potentiellement négatif de l'utilisation de ces médicaments et nous acceptons qu'il y ait une chance que cela nous arrive. L'utilisation d'un dispositif médical peut également être associée à des effets secondaires. L'utilisateur doit pouvoir juger par lui-même si les avantages de l'utilisation du dispositif l'emportent sur les risques, et le fabricant du dispositif doit donc fournir des informations sur les effets secondaires pour l'aider à prendre cette décision. Les autorités, chargées de protéger la santé publique, ont fait de l'évaluation des risques et de l'acceptabilité des effets secondaires un élément central de l'évaluation de la conformité des dispositifs médicaux. Pourtant, lorsque l'on entre dans les détails de la réglementation, ce sujet apparemment évident peut prêter à confusion.
Pour commencer, le règlement de l'UE sur les dispositifs médicaux (MDR) 2017/745 ne fournit pas de définition de l'effet secondaire. Pourtant, dans l'ensemble du texte, le terme apparaît 14 fois: une fois dans le contexte de la définition du terme "incident"; 2 fois dans le contexte des exigences générales en matière de sécurité et de performances (GSPR); 1 fois dans le contexte de l'évaluation clinique; 2 fois dans le contexte d'une investigation clinique; 2 fois dans le contexte des instructions d'utilisation; 3 fois dans le contexte de la surveillance post-commercialisation et du suivi clinique post-commercialisation (PMS/PMCF) et 3 fois dans le contexte de la déclaration d'incidents/de tendances.
Le MDR indique que l'apparition d'un effet secondaire indésirable est un type d'incident (article 2 [64]), que le risque d'effets secondaires doit être réduit au minimum et être acceptable lorsqu'il est mis en balance avec les bénéfices pour le patient (annexe I, section 8) et que les informations à cet égard doivent être fournies dans le mode d'emploi (annexe I, section 23.4g,t). Le MDR exige que l'industrie évalue les effets secondaires par le biais d'une évaluation clinique (article 61 ; annexe XIV, section 1a). Des investigations cliniques peuvent être menées pour "déterminer tout effet secondaire indésirable, dans des conditions normales d'utilisation du dispositif, et évaluer s'il constitue un risque acceptable au regard des bénéfices que le dispositif doit permettre d'obtenir" (article 62.1). Les effets secondaires connus doivent être surveillés et des efforts doivent être faits pour identifier les effets secondaires précédemment inconnus par le biais du PMS (annexe III, section 1.1a) et du PMCF (annexe XIV, section 6.1b). Les effets secondaires attendus ne doivent pas être signalés aux autorités compétentes, à moins qu'il n'y ait une augmentation statistiquement significative de la fréquence ou de la gravité de l'effet secondaire qui pourrait avoir un impact significatif sur l'analyse bénéfice-risque (articles 87 et 88).
Les diagrammes de Venn ci-dessous illustrent la relation entre les risques résiduels et les effets secondaires.
Vision parfaite du monde. Ce niveau de détail pourrait être suffisant pour la plupart des discussions.
D'autres possibilités existent en fonction de l'exhaustivité du développement du produit, de l'analyse des modes de défaillance et de leurs effets (FMEA), du rapport de gestion des risques (RMR), du rapport d'évaluation clinique (CER) et des spécificités du produit. Il semblerait qu'une partie ou la totalité des effets secondaires imprévus puissent être en dehors des risques résiduels connus.
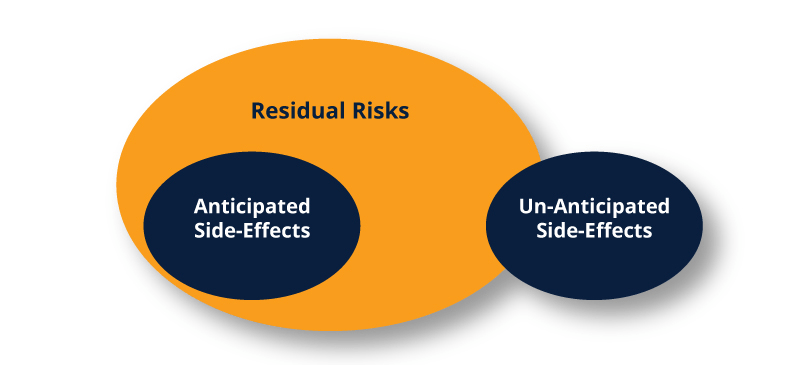
Dans ce cas, la documentation appropriée devrait être mise à jour pour les inclure. Les événements prévus dans le cadre des risques résiduels devraient augmenter et les événements imprévus devraient diminuer avec les mises à jour de le FMEA et du RMR.
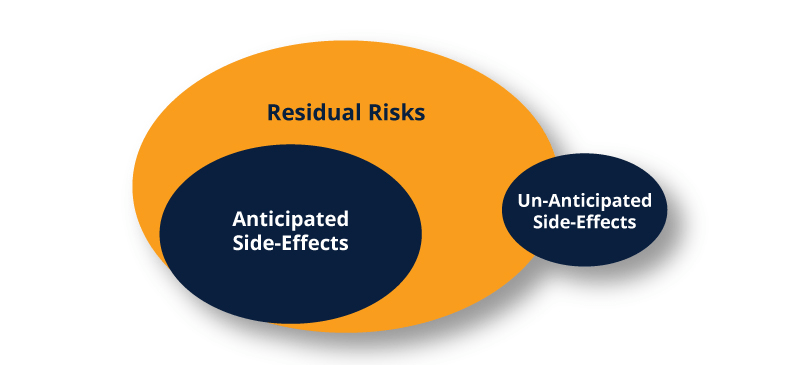
Pour la plupart des produits, il se peut qu'il n'y ait jamais d'effets secondaires imprévus, mais lorsqu'ils se produisent, ils font partie ou non des risques résiduels. Dans tous les cas, la documentation appropriée doit être mise à jour, ce qui nous ramène à la vision d'un monde parfait.

Toutes choses étant égales par ailleurs, il semblerait qu'un produit de technologie bien établie (WET) soit plus susceptible d'avoir surmonté la plupart des effets secondaires imprévus, alors qu'une nouvelle technologie n'ayant pas fait ses preuves aurait plus de chances d'avoir des effets imprévus.
Dans le cadre des études cliniques (avant la mise sur le marché et dans le cadre du PMCF), les effets secondaires jouent un rôle particulier. L'un des objectifs d'une investigation clinique peut être de recueillir des informations supplémentaires sur les effets secondaires connus et d'identifier des effets secondaires précédemment inconnus. Le plan d'investigation clinique (CIP), la brochure de l'investigateur (IB) et les demandes d'autorisation réglementaire et d'approbation éthique de l'investigation clinique doivent contenir des informations sur les risques prévisibles connus et les effets secondaires indésirables (ISO14155:2011/AC2011 et annexe XV du MDR). Au cours d'une étude clinique, la sécurité et les performances d'un dispositif sont étroitement observées. Tous les événements indésirables (EI) sont enregistrés. Un événement indésirable est défini comme "tout incident médical, maladie ou lésion non intentionnelle ou tout signe clinique indésirable, y compris un résultat de laboratoire anormal, survenu chez des sujets, des utilisateurs ou d'autres personnes dans le cadre d'une investigation clinique, qu'il soit lié ou non au dispositif expérimental" (article 2 [57]). Lorsqu'un EI est lié au dispositif expérimental, il est classé comme un effet indésirable du dispositif (ADE) (ISO14155:2011/AC2011, section 3.1).
Les ADEs peuvent être classés en fonction de leur gravité et de leur caractère anticipé ou non anticipé. Les ADEs sont imprévus lorsqu'ils n'ont pas été identifiés dans le cadre de la gestion des risques en cours. Lorsqu'ils entrent dans la catégorie des événements indésirables graves, ils doivent être signalés aux autorités des pays où l'étude a lieu (MDR et guidance MDCG 2020-10/1 "Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745"). La survenue d'effets graves imprévus sur le dispositif (USADE) peut suggérer que l'investigation clinique expose les sujets à un risque accru de préjudice par rapport à ce qui était prévu au préalable et peut avoir des conséquences sur la poursuite de l'étude (MDCG 2020-10 et ISO14155:2011/AC2011). C'est pourquoi vous devez inclure une liste complète des effets secondaires identifiés dans la documentation de l'étude clinique. Les données de l'étude alimenteront votre évaluation clinique et, le cas échéant, la gestion des risques pour le dispositif. Les données de l'étude sur les événements indésirables peuvent ainsi être utilisées pour étayer les preuves de la nature, de l'incidence ou de la gravité des effets secondaires associés à l'utilisation du dispositif.
Nous conseillons à nos clients de suivre une approche de gestion des risques et d'évaluer les informations provenant des rapports d'événements indésirables issus des investigations cliniques ou du suivi clinique après la mise sur le marché. Il faut considérer que les effets secondaires sont une forme de préjudice pour le patient (par exemple, une réaction allergique lors de l'exposition à un dispositif) et que le risque d'un tel préjudice est inhérent à l'utilisation du dispositif (c'est-à-dire que le risque subsiste après avoir pris des mesures d'atténuation du risque) et ne peut être réduit par des mesures de protection ou des informations sur la sécurité (la probabilité de survenue d'un préjudice et la gravité de celui-ci sont invariables). Tout risque clinique résiduel de ce type doit être signalé dans le dossier de gestion des risques et ajouté à la liste des effets secondaires dans les documents d'information accompagnant le dispositif.