Europäische Verordnung 2017/745 über Medizinprodukte
Was bedeutet MDR-Konformität?
Die europäische Verordnung 2017/745 über Medizinprodukte (MDR) ist ein Regelwerk, das die Herstellung und den Vertrieb von Medizinprodukten in Europa regelt.
Für Hersteller von Medizinprodukten, die ihre Produkte auf dem europäischen Markt vertreiben wollen, ist die Einhaltung der Verordnung verpflichtend. Hersteller müssen eine Reihe von MDR-konformen Systemen, Prozessen und Dokumenten einführen, um die Produktsicherheit kontinuierlich zu überwachen.
Unser Fahrplan zur Umsetzung der MDR:
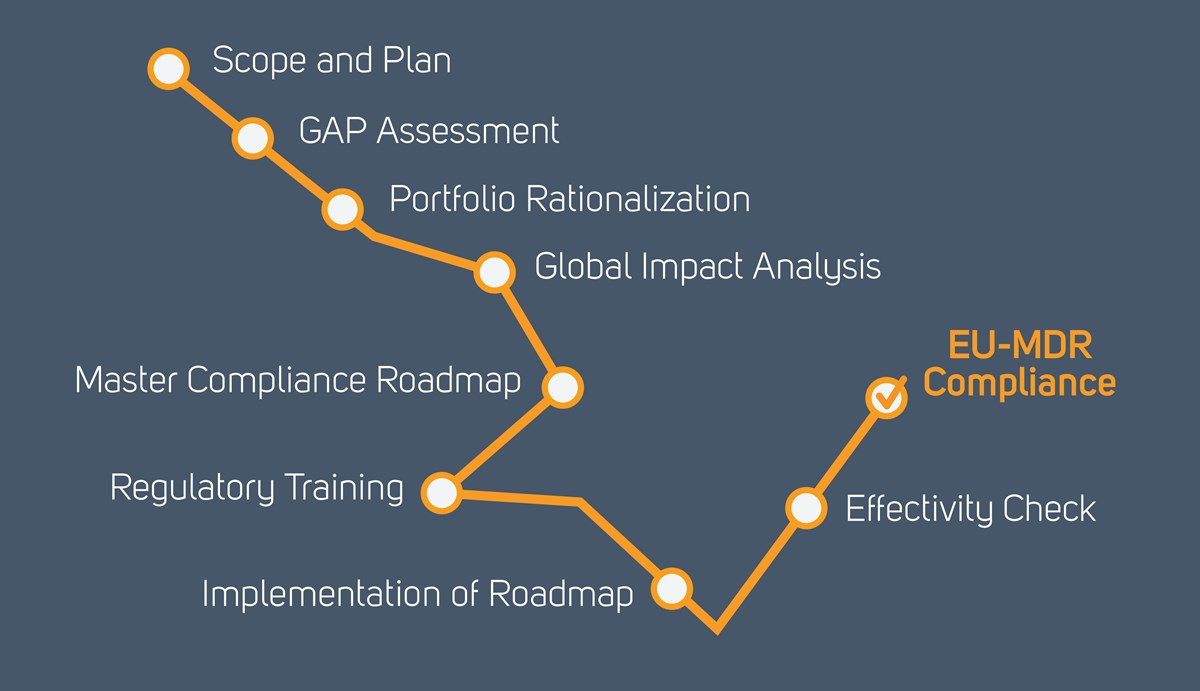
Von der MDD zur MDR
Die europäische Verordnung 2017/745 über Medizinprodukte (auch Medical Device Regulation/MDR) hat die Richtlinie 93/42/EWG über Medizinprodukte (auch MDD) als Rechtsvorschrift abgelöst. Sie legt die Anforderungen fest, die Hersteller erfüllen müssen, um Medizinprodukte in der Europäischen Union in Verkehr bringen zu können.
Alle Medizinproduktehersteller der Klassen I bis III müssen sich schnellstmöglich mit den neuen Anforderungen vertraut machen. Die Verordnung hat weitreichende Auswirkungen und betrifft alle Medizinprodukteklassen.
Im Vergleich zur MDD konzentriert sich die neue europäische MDR weniger auf die Phase vor der Zulassung von Medizinprodukten und fördert stattdessen einen lebenszyklusorientierten Ansatz bei der Regulierung von Medizinprodukten.
Die erfolgreiche Einhaltung der MDR wird durch die Anbringung des CE-Kennzeichens auf dem Produkt nachgewiesen. Hersteller von Produkten der Klasse I, mit Ausnahme von Produkten, die steril sind oder eine Messfunktion haben, können das CE-Zeichen nach Vorlage einer Konformitätserklärung selbst anbringen. Für alle anderen Medizinprodukteklassen darf die CE-Kennzeichnung nur angebracht werden, wenn eine Konformitätsbescheinigung einer Benannten Stelle nach behördlicher Prüfung gemäß den Bestimmungen des Kapitels IV der MDR vorliegt.
Sind Sie bereits MDR-konform?
Oder möchten Sie wissen, welche Auswirkungen die MDR auf Ihr Unternehmen hat? Wir bieten Ihnen eine kostenlose Konformitätsprüfung an.
MDR-Konformitätsprüfung
Wesentliche Änderungen der MDR gegenüber der MDD
Erweiterung des Produktumfangs - Die Definition von Medizinprodukten und aktiven implantierbaren Medizinprodukten, die unter die MDR fallen, wird erheblich erweitert. Sie umfasst nun auch Produkte ohne medizinische Zweckbestimmung.
Benennung einer "Verantwortlichen Person" - Medizinproduktehersteller müssen mindestens eine Person in ihrer Organisation benennen, die letztendlich für alle Aspekte der Einhaltung der Anforderungen der neuen Verordnung verantwortlich ist.
Implementierung einer einmaligen Produktkennung (UDI) - Die MDR schreibt die Verwendung von Mechanismen zur eindeutigen Produktidentifikation (Unique Device Identification - UDI) vor. Ziel dieser Anforderung ist es, Herstellern und Behörden die Möglichkeit zu geben, bestimmte Produkte durch die Lieferkette zu verfolgen.
Neuklassifizierung von Medizinprodukten nach Risiko, Kontaktdauer und Invasivität - Nach der MDR müssen Medizinproduktehersteller die aktualisierten Klassifizierungsregeln überprüfen und ihre technische Dokumentation entsprechend aktualisieren. Dabei sind die erhöhten klinischen Anforderungen an Medizinprodukte der Klasse III und implantierbare Medizinprodukte zu berücksichtigen.
Klinische Nachweise für Klasse III Medizinprodukte und implantierbare Medizinprodukte - Hersteller müssen klinische Studien durchführen, wenn keine ausreichenden klinischen Nachweise für die Sicherheit und Leistung ihrer Medizinprodukte vorliegen.
Klinische Bewertung von Medizinprodukten der Klassen IIa und IIb - Hersteller müssen ihre klinische Bewertung an die neue Formulierung der Verordnung bezüglich der Äquivalenz und des Verzichts auf eine klinische Prüfung anpassen.
Überwachung nach dem Inverkehrbringen - Mit der MDR wurde die Überwachung nach dem Inverkehrbringen durch die Benannten Stellen ausgeweitet.
Kein Bestandsschutz - Nach der MDR müssen alle derzeit zugelassenen Produkte nach den neuen Anforderungen neu zertifiziert werden.