

Our Clients
Trusted by the industry












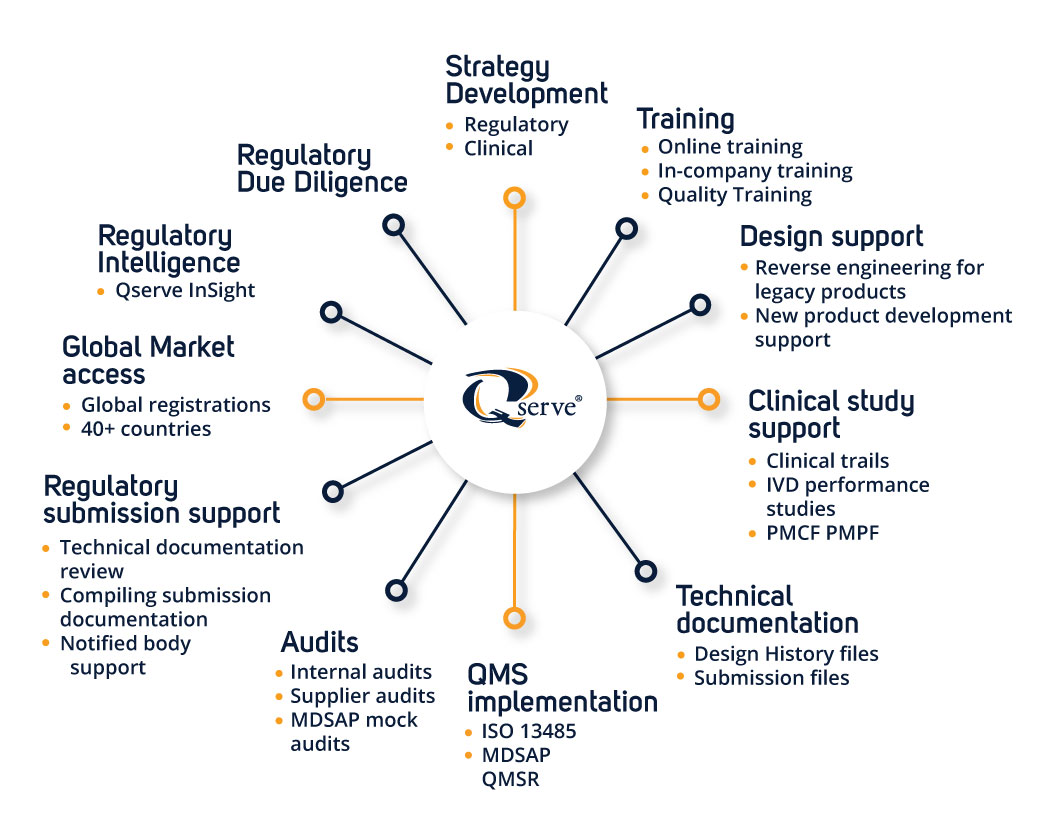
Product life cycle services
Ready to secure compliance and market access?
Whether you’re introducing a breakthrough innovation or managing the compliance of legacy devices, Qserve is here to guide you throughout the entire life cycle of your device.
Contact us today to discuss how we can support your needs.

CEP and CER are not the same document. Learn the critical difference between a Clinical Evaluation Plan and Report under EU MDR, and why it matters.
.png)
Discover how Qserve supported Jenscare Scientific in achieving EU MDR CE certification for the novel Class III LuX-Valve Plus™ cardiovascular device.
-3.png)
Discover the 5 top reasons Clinical Evaluation Reports fail MDR review, and what your team must do to get it right the first time.