

Regulatory compliance for Canada
Taiwan agent representation
Taiwan agent representation
Taiwan’s Ministry of Health and Welfare (MOHW) is the government ministry responsible for the country’s healthcare regulations. The MOHW has a broad mandate to improve healthcare quality, prevent and control infectious diseases, ensure food and drug safety, and facilitate technological development.
Within the MOHW, the Taiwan Food and Drug Administration (TFDA) manages the regulatory system for the safety and quality of food, drugs, medical devices, and cosmetics. The TFDA drafts and implements regulations, grants product registration and clinical trial approvals, monitor manufacturing and importation and conducts safety surveillance activities on health products.
In May 2021, Taiwan announced a new Medical Devices Act, which changes how medical device products are classified, groups products by risk and category, and adjusts registration fees. It also reinforces post-market surveillance of device safety and quality by mandating regulations for product recalls.

The Role of a Taiwan Agent in Medical Device Registration
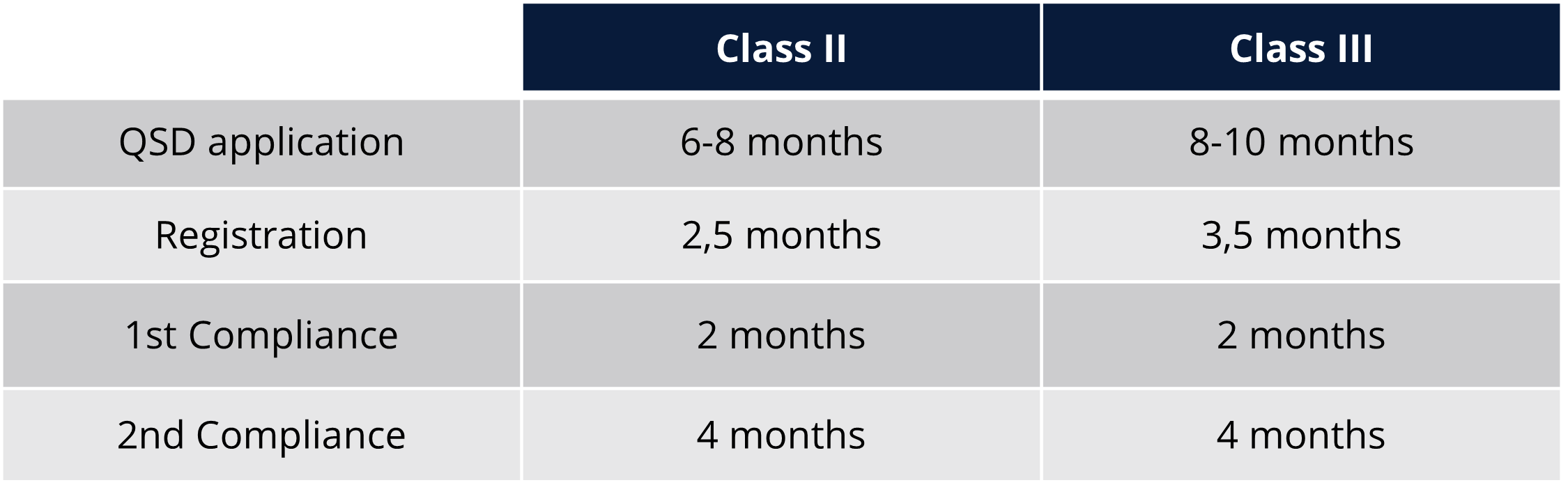
Regulatory timelines and fees for Taiwan
Class I (non-sterile)
- Class I medical devices without brand names (e.g., surgical instruments or power accessories) usually follow a simple self-declaration process in Taiwan.
- Timeline: The Class I registration approval process takes about 3 months.
- A list of Class I devices may be registered through the listing process outlined in Article 25 of the Medical Devices Act, effective October 1, 2021. The list contains 68 device codes.
Important links and guidance documents
.png)
Discover how Qserve supported Jenscare Scientific in achieving EU MDR CE certification for the novel Class III LuX-Valve Plus™ cardiovascular device.
-3.png)
Discover the 5 top reasons Clinical Evaluation Reports fail MDR review, and what your team must do to get it right the first time.
-2.png)
Predetermined Change Control Plans (PCCPs): A Quiet but Significant Shift in FDA Regulatory Strategy
Discover how FDA Predetermined Change Control Plans, PCCPs, enable proactive lifecycle change management and reduce future regulatory submissions.