

Involving submission specialists with up-to-date knowledge of the applicable regulations—the Medical Device Regulation (2017/745) and In Vitro Diagnostic Regulation (2017/746) in the EU, and the FDA in the US—will help guide you on which processes and documents are needed to ensure an efficient submission and approval process.
Important elements in the submission process are:
- Define your submission strategy in time. While we wait for EUDAMED to become fully operational, each country will have its own processes, submission methods, and requirements in place, which will lead to different timelines for creating the submission package and receiving approval for your project. These differences should be taken into account when defining which countries will be involved in your project.
- Ensure that submission specialists are involved, who have up-to-date knowledge of the requirements in the participating countries, and take their feedback into consideration while preparing the submission package.
- Ensure that the best possible quality package is submitted in the initial submission to avoid delays in the validation, review, and approval process. Please note that Requests for Information (RFIs) stop the review clock and can significantly delay approvals.
- Be aware of the processes and timelines regarding submissions for both Medical Device investigations and IVD studies. Some countries allow submissions directly to the authorities (Competent Authority/Health Authority) only as a notification under strict conditions (e.g. France), other countries require submission of the full packages to EC and CA/HA simultaneously (e.g. The Netherlands and France), and some require sequential submission—first to the EC and afterwards to the HA/CA (e.g. Spain and Italy).
- Required documentation can vary per country, but will contain at a minimum the Clinical Investigational Plan (CIP) for Medical Device studies and the Clinical Performance Study Plan (CPSP) for IVD studies, patient information and consent forms (when applicable), CVs of principal investigators including valid GCP training, cover letters per country, application forms per country layout, investigator brochure (IB), study plan synopsis, GDPR statement, and IFU when available. Additional requirements may apply in each country.
Combined trials
In the case of combined IVD/Pharma studies, submissions in the EU need to be made in the Clinical Trial Information System (CTIS) under the Clinical Trial Regulation (CTR, 536/2014) as well as in each country, according to the previously mentioned IVD Regulations. This adds complexity to this submission phase, since the two processes need to be aligned and, in some countries, combined into one submission. Version control and full approvals for all documents submitted to both CTIS and the local ECs and authorities are extremely important to ensure that full approvals have been received for all documents used in both the pharma and performance studies.
MDCG 2022-10: Q&A on the interface between CTR and IVDR, was issued to clarify how IVD assays should be used in Pharma Studies to meet IVDR requirements:
- It clarifies concept of a medical purpose of an assay in a clinical trial
- States that IVDs used in drug clinical trials MUST conform to the requirements of IVDR
- Clarifies the roles and responsibilities of the IVD manufacturer and pharma partner:
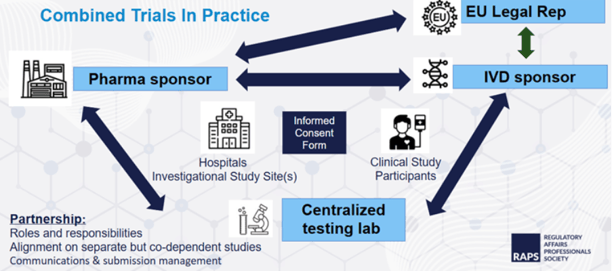
Whether a combined or standalone study or investigation is submitted, the complexity of the submission package, as well as timelines, will vary significantly by country. The time to receive approval per country can range from 2 months up to 1 year, depending on the number of RFIs, the complexity of the questions, the timeframe for submitting responses, and other factors.
Within Qserve, we have the expertise and knowledge to support you in these complex processes, helping you define the best strategy for your project, create submission documents of the highest quality, assist with the submission process, and follow up on RFIs when applicable.
Submissions regarding clinical investigations and performance studies can be complex, but you don’t have to navigate them alone. Contact Celien today for a free 30-minute consultation to receive personalized guidance and insights from a submission specialist’s perspective.
.png)
Plan your clinical investigation submission in Germany: ensure process clarity and MDR-aligned documents to avoid delays or questions from the EC and BfArM

Discover the differences between a Medical Device/IVD CRO and a Pharma CRO, and why understanding these concepts are essential when choosing a partner.

Global regulations are raising the bar for clinical data in medtech, driving a shift toward more robust pre- and post-market clinical strategies.