

Last Friday, the European Commission published a draft regulation to amend Annex VII of the EU MDR and the EU IVDR. In both laws, Annex VII sets out the requirements for the conformity assessments performed by the notified bodies (NBs) within the European regulatory frameworks for medical devices and IVD devices. These changes directly impact the timelines for manufacturers to bring their devices to market.
In this blog, we will focus on the impact on startups that are bringing their first device to the EU market. The focus of the regulation is on transparency regarding costs and timelines, making it easier to plan the conformity assessment part of a startup's go-to-market strategy.
While Annex VII is intended for notified bodies, it is essential to note that cooperation between notified bodies and manufacturers is bidirectional, and active participation is expected from manufacturers to ensure timely market access without incurring high costs.
A new requirement for NBs is that there should be a possibility in the quotation process for the applicant to give information that the company is, in fact, an SME. (less than 250 employees and less than 50 million Euro annual turnover).
The subsequent quotation must include the total costs required to perform the complete conformity assessment. Also, an estimate of potential additional costs shall be included.
The second part of the new regulation focuses on timelines that the notified body must keep:
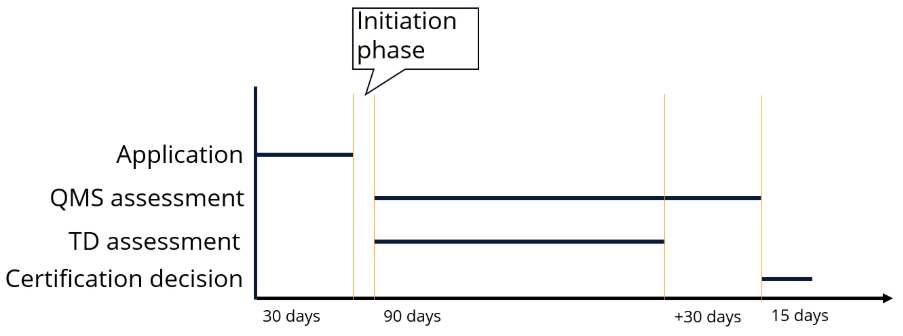
To initiate the conformity assessment with the notified body following the signing of the contract, an official application must be submitted, which includes the basic information already provided regarding the QMS and device.
After that, the NB initiates the QMS and TD assessment, which must happen in parallel. They will be interlinked, as the audit will need to consider some early review aspects from the TD along the way.
The notified body must use the shortest possible timeline, taking into account the type of device and company. This duration shall also be specified in writing.
The timelines mentioned in the regulation are one-sided and must be adhered to by the notified body. As mentioned earlier, the entire process is a bilateral cooperation. This means that on the manufacturer's side, some efforts are expected to address questions from the NB. The assessment timelines will be paused in these cases. How often this can happen is specified as follows:
- Application phase: 1 time
- QMS assessment: 3 times
- TD assessment: 3 times
Both parties shall agree upon the duration of these interruptions, and this agreement shall be put in writing as well.
|
Mandatory information by NB |
Agreements between the notified body and the manufacturer |
|
Increase in initially estimated costs, including justification of this increase |
Duration of interruptions of conformity assessment |
|
Interruption of conformity assessment due to EMA opinion, expert panel, regulatory authority, or EU reference lab |
Total timeline of assessment |
|
|
Extension of agreed-upon timelines |
As mentioned earlier, this document is still in draft and open for feedback. The official publication is expected to occur in Q2 2026 and will take effect three months later. You can adapt your planning to this if you are in a phase of starting your conformity assessment with the notified body. Part of your strategy should include one or more structured dialogues with the notified body you will be working with to ensure good communication and optimal use of the timelines.
With these newly defined timelines, the European market can once again compete on timelines with the US market and its regulatory framework.
Do you need help with your market access strategy or assistance during preparation for your conformity assessment procedure?
